Summary
PAX-D is a randomised placebo-controlled trial which aims to evaluate the efficacy and mechanism of pramipexole as add-on treatment for people with treatment resistant depression (TRD). Adults (age 18 and above) with TRD may be eligible for inclusion; full inclusion criteria can be found in the study protocol.
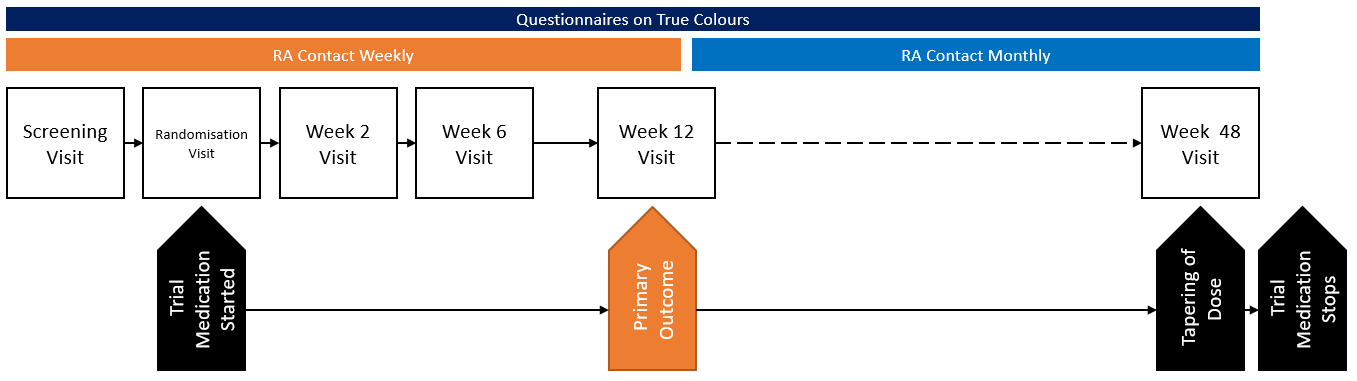
PAX-D will compare the effects of pramipexole with placebo when added to current antidepressant medication for people with TRD. The trial will look at effectiveness in the short- term (after 12 week’s treatment) and in the longer-term (48 weeks). The trial will also assess the adverse effects of pramipexole and explore patients’ experiences of taking it.
The primary outcome will be to measure improvement (change from baseline to week 12) of depressive symptoms measured on the Quick Inventory of Depressive Symptomatology, self-report version (QIDS-SR16).
The study is sponsored by the University of Oxford, and is run at the Department of Psychiatry, University of Oxford. The study is funded by the National Institute of Health Research (NIHR), Efficacy and Mechanism Evaluation programme (ref: 16/127/17). It has been approved by South West – Central Bristol Research Ethics Committee (ref: 19/SW/0216).
Study Population
Adults over the age of 18 years with treatment resistant depression (defined as a lack of response to at least two antidepressants at therapeutic doses in the current episode).
Inclusion Criteria
For entry into the trial:
- Willing and able to give informed consent to participate in the trial
- Males or females aged 18 years or over
- Diagnosis of DSM-V major depression based on the affective disorder sections of the Mini International Neuropsychiatric Interview (MINI). N.b. comorbid anxiety disorder is not an exclusion criterion
- Quick Inventory of Depressive Symptomatology self-report version (QIDS-SR16) score >10 (moderate, severe or very severe depression)
- Currently taking an antidepressant medication
- A lack of response to at least two antidepressants at therapeutic doses (based on Maudsley Prescribing Guidelines and/or British National Formulary) in the current episode
- An indication for a change in treatment
- Willing to continue antidepressant treatment
- Women of child-bearing potential (WOCBP) only – a negative urine pregnancy test result
Exclusion Criteria
- Clinical diagnosis of current or previous psychosis (including psychotic depression), bipolar disorder or Parkinson’s Disease
- Currently taking an antipsychotic medication
- Clinically significant current or previous impulse control difficulties
- Serious suicide or homicide risk
- Current treatment with any medication known to interfere with pramipexole metabolism including cimetidine, memantine and methyldopa
- Contraindications to pramipexole including history of or current treatment for eye disease (excluding near or long-sightedness), significant, symptomatic cardiovascular or renal disease or significant, symptomatic orthostatic hypotension
- Previous course of pramipexole (>2 weeks)
- Untreated or unstable medical condition which, in the judgement of the investigator, could interfere with the safety of receiving pramipexole or ability to complete the trial
- Female and pregnant, breast-feeding or planning pregnancy
- WOCBP not willing to use effective contraception
Study Arms
Participants are randomised either treatment with pramipexole or a matched placebo.
Primary Objective
To compare the efficacy of pramipexole and placebo at 12 weeks post-randomisation.
Secondary Objectives
- To compare the tolerability and safety of pramipexole and placebo during the 48 week treatment phase
- To compare the effect of pramipexole and placebo on reward sensitivity
- To test the degree to which change in reward sensitivity mediates the 12 week response to pramipexole of both depressive and specifically anhedonic symptoms
- To compare the extent to which an increase in reward sensitivity predicts therapeutic response
- To explore the extent to which reward sensitivity at baseline predicts therapeutic response
- To explore the extent to which level of anhedonia at baseline predicts therapeutic response
- To compare the effect of pramipexole and placebo on the trajectory of symptoms of depression
- To compare the effect of pramipexole and placebo on response and remission rates, using the QIDS-SR16, at twelve weeks
- To compare the impact of pramipexole and placebo on symptoms of anhedonia, anxiety and clinician rated depression
- To compare the impact of pramipexole and placebo on functional outcome over the 48 weeks of treatment
- To determine the impact on quality of life and wellbeing of pramipexole relative to placebo over 48 weeks
- To examine the health / social care and broader societal costs of patients relative to placebo over 48 weeks